
under construction; last updated December 1, 2003
> Home > Computational Epidemiology Course > Lecture 13
Analysis of HIV infectivity
In this lecture, I'll discuss some ways to use R to analyze data
from the San Francisco Young Men's Health Study1. We'll demonstrate a few simple programming techniques, and use a
modified version of the real data (i.e. fictitious data!) for
confidentiality. Some of the results in this web page (using the
real data, of course) are to appear shortly2
and the background literature, actual
scientific results, and discussion are to be found in that reference.
The problem of how infectious HIV is is not a simple one. Here, we'll
focus on one approach, that of per-partnership infectivity3. By infectivity, we mean the probability that an
individual is infected during a partnership given that the partner
is infected.
Now, what constitutes a partnership? We are going to restrict our
attention to partnerships in which the subject undergoes receptive
anal intercourse one or more times. We'll further distinguish
between partnerships in which the person reports that condoms were
always used (protected partnerships), and partnerships
for which condoms were not always used (unprotected partnerships).
What we wanted to know was whether there was any evidence that the
infectivity had declined after the widespread use of highly active
antiretroviral therapy began in 1996. Of course, the causal inference
is entirely circumstantial, in that we have no control group and no
assessment of the exposure of partners of men in the study to HAART.
And moreover, new subjects weren't recruited each year into the study,
leaving open the possibility of bias due to aging and differential
loss to follow-up.
The San Francisco Young Men's Health Study itself
is a multistage probability
sample (which began in 1992) of single men aged 18-29 who resided in
21 census tracts with the highest cumulative AIDS
incidence1.
It included 428 homosexual men at baseline and then enrolled an additional 622 referral subjects at the next follow-up time.
At each approximately yearly study visit, subjects were
tested for HIV antibodies
and were asked, with respect to the previous 12 months,
for their (1) number of sex partners,
(2) number of sex partners under 30 years of age, (3)
number of receptive anal intercourse partners,
and (4)
number of receptive anal intercourse partners with whom condoms were always used.
The last four study visits
used consistent questions about behavior and provided two
periods at risk both before (4/94 to 9/95; and 9/95 to 11/96) and after
(11/96 to 9/97; and 9/97 to 3/99)
the introduction of HAART in San Francisco and were used in the
analysis.
One problem we will discuss is that the subjects were asked about their
sexual practices in the last 12 months, but more or less than 12 months
elapsed since the last HIV test in many cases. Another problem is that
as much as 3 months may sometimes elapse between infection and
seroconversion (though median seroconversion times are much less than
3 months).
Data format
We first want to talk a bit about data formats and importing. R
can import data in both fixed and delimited formats.
Here, we have four data sets, one for each of the four study
periods we decided to analyze. There are many different ways to
represent it in the computer; we chose to simply represent each
data set by four different data frames. It would also have been
possible to merge them into a single data frame with more variables.
Here is what the data file looked like (remember, these are
false data points):
3 0 NA 0 NA NA "500555a01" 0.95
3 0 1 1 1 0 "500555s01" 1.5
3 0 5 6 3 3 "199691d01" 1.3
|
The variables consisted of the study period, the HIV status (0 for
uninfected, 1 for infected), the number of partners under 30 (all
kinds of partnerships), the number of partners total, the number
of RAI partnerships, the number of RAI partnerships for which
condoms were always used, a study identifier (a string), and finally
the fraction of one year that was elapsed since the last HIV test
(neglecting seroconversion delays). So for the last row of data
I showed you, you see study period 3, the person did not seroconvert,
they had 5 out of their 6 partnerships with men 30 or younger, they
had RAI with three of these men and used condoms all the time for each
of these three partners, and 1.3 years had passed since their last
HIV test.
It's important to realize that for each subject in our analysis, we
included only subjects who were known to be HIV negative at the
beginning.
Several things should be noted about these data. First, each record
(all the variables from one subject) are located on a single line, and
the values are separated by spaces. This is called "whitespace
separated". Second, missing values have already been coded as
NA, which as you know is the missing value code that R and S
use. Different statistics programs use different codes, and moreover,
survey centers may use still different codes (and often in the
same data set); for instance, a value
of 9 may denote a missing value for some variables, a dot (.) may
denote a missing value for others, and perhaps a 999 for others. And
sometimes you see several different missing value codes depending on
exactly what happened, that may distinguish various kinds of nonresponse. So handling missing values gracefully, and more generally cleaning the
data, is not at all a trivial matter or one to be taken for granted.
Before
the data were loaded in to R, all these were recoded to NA, though we
could easily have done the recoding in R. Third, we chose to use no
header (i.e., there is no line at the top containing variable names).
Because there were 4 data sets, we chose to write a simple function
to read the data in, and then apply it to four files:
> getdata <- function(fname) {
+ thedata <- read.table(fname,header=FALSE);
+ vars <- c("studywave","hivstatus","nyoung","ntot","nrai","nrai.prot","id","frac.yrs.since.test")
+ dimnames(thedata)<-list(thedata[,7],vars)
+ thedata
+ }
|
How does this work? First, the argument to the function is
a file name, where the data is located. We use the function
read.table to actually read in the
data. Then we chose to directly assign the variable names (vars) as the column names of the data frame thedata by assigning to
dimnames.
Let's spend some time on this. Let's suppose that a little data
set containing only the lines I showed you is filed as td1
(test data 1). Let's read it in:
> # continuing above example
> testdata <- getdata("td1")
> testdata
studywave hivstatus nyoung ntot nrai nrai.prot id
500555a01 3 0 NA 0 NA NA 500555a01
500555s01 3 0 1 1 0 0 500555s01
199691d01 3 0 3 4 2 2 199691d01
frac.yrs.since.test
500555a01 0.95
500555s01 1.50
199691d01 1.30
|
Here, it printed the row names on the side, and we chose to use the
id column as the row names. We then see the column names on
the top, studywave, hivstatus, etc. It wrapped
around and made a separate series of lines for frac.yrs.since.test. We could perfectly well erase the id column now if we wished, since it is the same as the row names.
> #continuing...
> is.data.frame(testdata)
[1] TRUE
> dimnames(testdata)
[[1]]
[1] "500555a01" "500555s01" "199691d01"
[[2]]
[1] "studywave" "hivstatus" "nyoung"
[4] "ntot" "nrai" "nrai.prot"
[7] "id" "frac.yrs.since.test"
|
Note that the result of applying the dimnames function to
testdata was a list with two parts: the first part being
the row names, and the second the column names (each part being
a vector). Importantly, it is possible to actually assign values to
dimnames, as you saw us do inside the getdata function.
For instance, should we choose to change the names to something
different, we could do so:
> #continuing...
> dimnames(testdata) <- list(c("person1","person2","person3"),
c("var1","var2","var3","var4","var5","var6","var7","var8"))
> testdata
var1 var2 var3 var4 var5 var6 var7 var8
person1 3 0 NA 0 NA NA 500555a01 0.95
person2 3 0 1 1 0 0 500555s01 1.50
person3 3 0 3 4 2 2 199691d01 1.30
|
Now when the data are displayed, you see the new data names for the
rows and columns. (These new names aren't very good, but they work
the same way.) You can even change just the row names:
> #continuing...
> dimnames(testdata)[[1]] <- c("aa","bb","cc")
> testdata
var1 var2 var3 var4 var5 var6 var7 var8
aa 3 0 NA 0 NA NA 500555a01 0.95
bb 3 0 1 1 0 0 500555s01 1.50
cc 3 0 3 4 2 2 199691d01 1.30
|
Exercise: How would you change just the column names without
affecting the row names?
Next semester we will learn how to make functions of our own that
can appear on the left hand side of assignment.
Let's now return to where we were:
> getdata <- function(fname) {
+ thedata <- read.table(fname,header=FALSE);
+ vars <- c("studywave","hivstatus","nyoung","ntot","nrai","nrai.prot","id","frac.yrs.since.test")
+ dimnames(thedata)<-list(thedata[,7],vars)
+ thedata
+ }
> testdata <- getdata("td1")
|
Notice that we hard-coded the number seven as the location of the
id field. Notice what can go wrong now: first, it is possible
that the format of the data could change, so that the variable names
we have listed in vars might not correspond to the data set
any more; the data set is not self-documenting. And just as bad, we
have a separate reference to 7 which has to be id. These
two features make this program brittle: we can't change the
format of the data without changing the program in two places:
if we add an extra variable in column 2 then we had better change
vars and we had better make that 7 into an 8 or we have an
error.
So normally it is better to have the variable names associated with
the data set itself as a header:
studywave hivstatus nyoung ntot nrai nrai.prot id frac.yrs.since.test
3 0 NA 0 NA NA "500555a01" 0.95
3 0 1 1 1 0 "500555s01" 1.5
3 0 5 6 3 3 "199691d01" 1.3
|
This is much better. Now, though, we will need a new getdata:
> getdata <- function(fname) {
+ thedata <- read.table(fname,header=TRUE);
+ dimnames(thedata)[[1]]<-thedata[,"id"]
+ thedata
+ }
> testdata <- getdata("td1")
|
This works fine. Here we had to use header=TRUE
because the first line was a header. We also directly referred to
id instead of column 7, so that this line works whereever the
id variable is. Both this version and the previous one are
"right", but this version is a little better since this version
requires less work to stay right if the data format changes.
Of course, as always, "to err is human, but to really foul things up
requires a computer" (as the saying goes). Here are a few bloopers
that might happen at this stage:
> getdata <- function(fname) {
+ thedata <- read.table(fname,header=TRUE);
+ dimnames(thedata)[[1]]<-list(thedata[,"id"])
+ # Error--this list component should be a vector of row names,
+ # not a list!
+ thedata
+ }
> testdata <- getdata("td1")
Error in "dimnames<-.data.frame"(*tmp*, value = list(thedata[, "id"])) :
invalid dimnames given for data frame
|
My, what a sorehead...it really wants a vector and nothing else will
do. (By the way, the error message gives you a clue as to what
is really happening...there seems to be a function called something
like dimnames<-.data.frame that is actually being called
to do the operation that looks like assigning to a function value
in dimnames(testdata) <- .... Stay tuned next semester.)
How about this?
> getdata <- function(fname) {
+ thedata <- read.table(fname,header=TRUE);
+ dimnames(thedata)[[2]]<-thedata[,"id"]
+ # Error--we're assigning to the columns, not the rows, and the
+ # length is worng
+ thedata
+ }
> testdata <- getdata("td1")
Error in "dimnames<-.data.frame"(*tmp*, value = list(thedata[, "id"])) :
invalid dimnames given for data frame
|
Well, that does not work either. If you're assigning the first
component of dimnames, then the length of the vector has to
match the number of rows. And if you're assigning the second
component of dimnames, then the length of the vector has to
match the number of columns. Or else it just won't do at all.
Of course if you have the same number of rows as columns, then you
can indeed get them transposed: the computer can't read your mind.
(And I for one don't want there to ever be a do.what.I.mean function.)
Here is another mistake you'll see:
> getdata <- function(fname) {
+ thedata <- read.table(fname,header=FALSE);
+ dimnames(thedata)[[2]]<-thedata[,"id"]
+ # Error--we said no header, so the header will be treated as
+ # data.
+ thedata
+ }
> testdata <- getdata("td1")
Error in "dimnames<-.data.frame"(*tmp*, value = thedata[, "id"]) :
invalid dimnames given for data frame
|
Luckily, we get the same old error message. But this time it
happens because thedata[,"id"] does not actually exist. If
we had not had the line dimnames(thedata)... in the
function, this actually would have read in the data wrong and it would
have coerced everything to character data. Of course, another mistake
would be to have no header in the file, but specify to R that there
is a header; in this case, the first line of your data becomes the
variable names...not something to brag about.
Ok, so we have a better version of getdata, but it still
is not good enough. Now we let the data file take care of the
variable names, but we still need to be sure that the variable
names in the data are what the program expects. After all, what if
these data are being prepared for you by a data manager who gets the
variable names worng, or who uses a capital letter when you want
lowercase, or something else. We'd better check and make sure we
have everything we want.
Suppose the data manager sent us the data set in a slightly
different format, with an extra variable Index at the
beginning, and with the variable studywave spelled as
Studywave:
Index Studywave hivstatus nyoung ntot nrai nrai.prot id frac.yrs.since.test
1 3 0 NA 0 NA NA "500555a01" 0.95
2 3 0 1 1 0 0 "500555s01" 1.5
3 3 0 3 4 2 2 "199691d01" 1.3
|
Now there's nothing really wrong with this; it's just a little
different. But Studywave is not the same name as studywave as far as R is concerned, so we'd better catch this:
> getdata <- function(fname,required=c()) {
+ thedata <- read.table(fname,header=TRUE);
+ dimnames(thedata)[[1]]<-thedata[,"id"]
+ nreq <- length(required)
+ if (nreq>0) {
+ colnames <- dimnames(thedata)[[1]]
+ for (ii in required) {
+ if (!(any(colnames==ii))) {
+ stop(paste("name",ii,"missing in data set"))
+ }
+ }
+ }
+ thedata
+ }
> testdata <- getdata("td1",c("studywave","hivstatus","nyoung","ntot","nrai","nrai.prot","id","frac.yrs.since.test"))
Error in getdata("td1", c("studywave", "hivstatus", "nyoung", "ntot", :
name studywave missing in data set
|
That's better. Now the program will at least check if the variables
it wants are there. Notice that the way it works is that if we
specify the second argument required to be something, then
it will check and make sure each of those items are variables; otherwise
it defaults to c() (an empty vector), and the check is never
performed. The check itself is performed by looping through each
required variable name, comparing each of them to every required variable name, and stopping if
a variable name is not found. Inside the loop, ii
takes each value in required in turn, one value for each
loop iteration. The operation colnames==ii
produces a boolean (logical) vector, with TRUE in the position of
colnames where ii is and FALSE everywhere else; if
there is a TRUE somewhere, then ii is in
colnames (that required variable is in the data set),
any(colnames==ii) is TRUE, and everything is fine. But if
this is not true, i.e. if !(any(colnames==ii) is TRUE, then
the required variable ii did NOT occur in the data set, and
so we stop and give an error message. If we get through all the
required variables without stopping, then all the required
variables are present and we can proceed.
One other thing that was done
to remove all data points that were inconsistent, in the sense of
having negative numbers of partners, missing numbers of partners,
or more protected partnerships than total partnerships. There are
many ways to do this; I chose to write a simple function
> inconsistent.1 <- function(datum) {
+ sa<-datum["sa"]
+ sap<-datum["sap"]
+ tot<-datum["tot"]
+ sy<-datum["sy"]
+ ans <- (is.na(sa) | is.na(sap) | sa<0 | sap<0 | sa |
and apply it to the data frame:
> keep.all <- function(datum0) {
+ ns<-dim(datum0)[1]
+ boolout <- rep(FALSE,ns)
+ for (ii in 1:ns){
+ boolout[ii] <- as.vector( !(inconsistent.1(datum0[ii,])) )
+ }
+ boolout
+ }
|
to produce a vector of boolean values.
Then this function could be applied:
data3to4final <- data3to4[keep.all(data3to4),] ;
dimnames(data3to4final)[[1]] <- data3to4final[,"id"];
|
This is not particularly efficient, but it only has to be done
once at the beginning of the analysis.
Fraction of partners under 30
We wanted to use the fraction of partners under 30 to see if
age mixing could be related to whatever findings we made.
Unfortunately the data weren't collected at the last study period
(which was actually the baseline for another study). Also, for
people who did not report the ages of their partners, we wanted to
use the overall fraction for that study period to impute it (what
other imputation comes to mind?) So we wanted to be able to
calculate the fraction of partners in each study period under 30.
Here is one (admittedly pedestrian) way to do it:
> gen.overall.youngfrac <- function(dataset) {
+ totptrs <- 0
+ totyptrs <- 0
+ for (ii in 1:(dim(dataset)[1])) {
+ tot<-dataset[ii,]$tot
+ sy<-dataset[ii,]$sy
+ if (is.na(tot) | is.na(sy)) {tot<-0;sy<-0}
+ totptrs <- totptrs+tot
+ totyptrs <- totyptrs+sy
+ }
+ totyptrs/totptrs
+ }
|
As a homework problem, how would you do this in a more vectorized way?
Then, we only need to type
eprev3to4 <- gen.overall.youngfrac(data3to4final)
|
for example, to deal with the data set we're calling
data3to4final.
Other data formats
It is common to get comma separated versions of data, so you may have
had a data set like this:
studywave,hivstatus,nyoung,ntot,nrai,nrai.prot,id,frac.yrs.since.test
3,0,NA,0,NA,NA,500555a01,0.95
3,0,1,1,0,0,500555s01,1.5
3,0,3,4,2,2,199691d01,1.3
|
In that case, instead of using read.table, we would have
used read.csv.
By the way, for some reason, it's not uncommon to get CSV files that
might look something like this:
studywave,hivstatus,nyoung,ntot,nrai,nrai.prot,id,frac.yrs.since.test,,,,,,,,,, ,
3,0,NA,0,NA,NA,500555a01,0.95
3,0,1,1,0,0,500555s01,1.5
3,0,3,4,2,2,199691d01,1.3
|
Notice all the null fields at the end. This seems to happen when people
export you spreadsheets. But it can certainly cause a problem in
reading the data:
> testdata <- getdata("td1")
studywave hivstatus nyoung ntot nrai nrai.prot id
500555a01 3 0 NA 0 NA NA 500555a01
500555s01 3 0 1 1 0 0 500555s01
199691d01 3 0 3 4 2 2 199691d01
frac.yrs.since.test X X X X X X X X X X
500555a01 0.95 NA NA NA NA NA NA NA NA NA NA
500555s01 1.50 NA NA NA NA NA NA NA NA NA NA
199691d01 1.30 NA NA NA NA NA NA NA NA NA NA
X
500555a01 NA
500555s01 NA
199691d01 NA
|
Here, it just gave us a lot of garbage variables named X. But if you
got the commas somewhere else, you might have seen a different error:
studywave,hivstatus,nyoung,ntot,nrai,nrai.prot,id,frac.yrs.since.test
3,0,NA,0,NA,NA,500555a01,0.95,,,,
3,0,1,1,0,0,500555s01,1.5
3,0,3,4,2,2,199691d01,1.3
|
So now you get:
> testdata <- getdata("td1")
Error in read.table(file = file, header = header, sep = sep, quote = quote, :
more columns than column names
|
Not surprising, since the machine thinks there is a record between
each comma.
Other problems that come up from time to time. For instance, suppose
you had this file:
"Name","Diagnosis","Date"
'Doe, John',"HCV","11/11/1999"
'Roe, Jane',"HCV","11/12/1999"
'Zygill',"Pteromic Plague","11/15/1999"
|
and you tried to read it in:
> z <- read.csv("td2",header=TRUE)
> z
Name Diagnosis Date
'Doe John' HCV 11/11/1999
'Roe Jane' HCV 11/12/1999
'Zygill' Pteromic Plague 11/15/1999
> z[,"Diagnosis"]
[1] HCV HCV 11/15/1999
Levels: 11/15/1999 HCV
|
This actually is completely incorrect, because it has gotten the
data mixed up into the worng columns. This happened because the
quote character in the data should have been ", not '. You can
change the quote character, however, as the manual page for
read.csv will show you. (...apologies to James Alan
Gardner, from whose novel Vigilant we got the extraterrestrial
disease Pteromic Plague). By the way, that last data item
was read in as a factor, an important data type used in many
statistical analyses, and about which we will say more.
There is one more thing we need to at least briefly mention: the
fixed format data file. It is quite common in the real world to
transfer data in fixed format, where each column has a meaning. Such
data formats are common when dealing with very large data sets.
Suppose you have a data file with four variables, and let's call
them A, B, C, and D to keep this short. Variable A is in column
1, B in column 2, C in column 3, and D in columns 4-5. For
instance, suppose the file td3 contains:
Now we want the A value for the third record to be missing, and the
D value for the third record to be 2. We can read these data in
to R, as follows:
> thedata <- read.fwf("td3",widths=c(1,1,1,2))
> thedata
V1 V2 V3 V4
1 2 3 A 89
2 2 4 Z 98
3 NA 3 F 2
> dimnames(thedata)
[[1]]
[1] "1" "2" "3"
[[2]]
[1] "V1" "V2" "V3" "V4"
|
You can also see the default variable and row names it chose. We used
read.fwf to read in the fixed width format
data; learn more about this important function from the manual page.
So there are many things that can go wrong in data importation in
any program, and this can happen to you as an R user. (The oddest
that happened to me was when text identifiers of the form
100200E09 for instance were misread as floating point data, becoming
1.002e+14.)
With care and
knowledge of the program, and defensive programming, you can
eliminate many such errors.
The model
Since we're considering the infectivity
 to be the same for each person and for each partnership, we could
begin to compute the infection probability as follows. For the
simple model, let's neglect the age of the reported partnerships
for now. Each subject reported, for the last 12 months, the number of protected
partnerships and the number of unprotected partnerships (remember
the subject reported RAI (receptive anal intercourse) for these
partnerships). Let's call the number of unprotected partnerships
n and the number of protected partnerships m.
to be the same for each person and for each partnership, we could
begin to compute the infection probability as follows. For the
simple model, let's neglect the age of the reported partnerships
for now. Each subject reported, for the last 12 months, the number of protected
partnerships and the number of unprotected partnerships (remember
the subject reported RAI (receptive anal intercourse) for these
partnerships). Let's call the number of unprotected partnerships
n and the number of protected partnerships m.
Now, to begin with, suppose that the last test was conducted not quite
a year ago. If it was conducted 9 months ago, then 0.75 of a year
has elapsed since the test; if it was conducted 11 months ago, then
11/12=0.917 of a year elapsed since the test. So let
 be the fraction of a year that has elapsed; thus, we're assuming
that
is less than 1.
be the fraction of a year that has elapsed; thus, we're assuming
that
is less than 1.
Now, we're going to neglect the few to several weeks that elapse between
infection and seroconversion in the model to follow. We believe this to
be not large compared to one year; it will introduce some misclassification
into the model.
Assuming the partners to be spread over the year at random, we could
assume that a given reported partnership has a
chance of having happened since the test. So what is the chance of
seroconverting? For one reported partnership, the chance of seroconverting due to it is the chance it happened since the last test, times the chance
the partner is infected, times the infectivity. If we denote the
chance the partner is infected by p, then the chance of
infection for that reported partner is
p. So
the chance of not being infected for that partnership is
1-p.
Then the chance of not being infected for all the unprotected partnerships (of which there are n) is
(1-p)n.
Now, what to do about the m protected partnerships? Let's say
that the infectivity is smaller if the person reports always using
condoms. So we'll multiply down the infectivity by a factor
 which is less than or equal to 1. So if
is 0.05, then you only have 5% the chance per partnership with an infected
person of becoming infected, etc. If we use the same simple assumptions
as above, then we get the chance of not being infected for all
the protected partnerships as
(1-p)m.
which is less than or equal to 1. So if
is 0.05, then you only have 5% the chance per partnership with an infected
person of becoming infected, etc. If we use the same simple assumptions
as above, then we get the chance of not being infected for all
the protected partnerships as
(1-p)m.
So the overall chance of not being infected is
(1-p)n × (1-p)m,
and the chance of being infected is
1-(1-p)n × (1-p)m.
What do we do if more than one year has elapsed since the test? For
instance, suppose it's been 18 months since the test; then the
actual risk period is 6 months (0.5 yr) longer than the time
since the test. We call this period of excess risk
 .
If the person says they had (say) 6 partners in the 12 months, you
might think that the person might be having a partner every 2 months.
So you might think that there were really an extra 3 partners since the
test, and we are going to have to come up with some sort of simple
adjustment for this. Now as it happens, just this simplest sort of
imputation worked quite well, but we actually did slightly more
involved imputation. We modeled the chance of not being infected
during the 12 months just the same way as above (with
=1). Then we modeled the chance of escaping infection in the remaining
period (the time beyond 12 months, since the test) as
exp(-pn)
for unprotected partnerships, and
exp(-pm)
for protected partnerships. This was based on assuming that the
observed rate of (self-reported) partnerships in 12 months is our
best estimate of the rate that partnerships occur in the remaining
risk period, and that given this rate, the partnerships occurred
according to the Poisson distribution (each small interval of time
had the same small chance of a partnership occurring in it). (We tried
other more complex imputation schemes and found that it made
essentially no difference.)
.
If the person says they had (say) 6 partners in the 12 months, you
might think that the person might be having a partner every 2 months.
So you might think that there were really an extra 3 partners since the
test, and we are going to have to come up with some sort of simple
adjustment for this. Now as it happens, just this simplest sort of
imputation worked quite well, but we actually did slightly more
involved imputation. We modeled the chance of not being infected
during the 12 months just the same way as above (with
=1). Then we modeled the chance of escaping infection in the remaining
period (the time beyond 12 months, since the test) as
exp(-pn)
for unprotected partnerships, and
exp(-pm)
for protected partnerships. This was based on assuming that the
observed rate of (self-reported) partnerships in 12 months is our
best estimate of the rate that partnerships occur in the remaining
risk period, and that given this rate, the partnerships occurred
according to the Poisson distribution (each small interval of time
had the same small chance of a partnership occurring in it). (We tried
other more complex imputation schemes and found that it made
essentially no difference.)
So based on this simple model, we could compute the likelihood function
of the data. Because (in part) of the small numbers involved in such
probabilities, we computed the logarithm of the likelihood.
It is important to realize that in this model, prevalence and
infectivity are completely nonidentifiable. In the paper, we discuss
the issues involved in trying to assume reasonable bounds on the
possible prevalence change over the study period and what this means
in terms of statistical inference on the value of the infectivity.
So let's construct a loglikelihood function. In will go a data
set and the parameters, and out will come the log-likelihood.
The parameters we're going to solve for (the infectivities for each
of the four study periods, and the condom protection value theta ()) will be located in the argument inpars. The first four
elements of the vector inpars are the four period-specific
infectivities, and the fifth element is theta. The second
argument dataset is the data set we're looking at, the fourth
argument eprev is a vector of study period specific
fractions of partners under 30. That leaves the third argument
which we called inprev.z, which is a matrix of period and
age-group specific prevalences of HIV infection, as shown.
> # several potential age-group/period specific prevalences
> # row is age group, column is period (3 is first period of
> # our analysis
> inprev.1 <- rbind(c(0.23,0.23,0.23,0.23,0.23,0.23),
+ c(0.23,0.23,0.23,0.23,0.23,0.23))
> inprev.2 <- rbind(c(0.23,0.23,0.23,0.22,0.21,0.20),
+ c(0.23,0.23,0.23,0.22,0.21,0.20))
> simlik.1<-function(inpars,dataset,inprev.z,eprev) {
+ wave <- as.integer(dataset[1,"studywave"])
+ beta <- inpars[wave-2] ;
+ theta <- inpars[5] ;
+ phi<-pmin(1,dataset[,"frac.yrs.since.test"])
+ tau<-pmax(1,as.vector(dataset[,"frac.yrs.since.test"]))-1
+ sy <- as.vector(dataset[,"nyoung"])
+ tot <- as.vector(dataset[,"ntot"])
+ yfrac <- sy/tot
+ yfrac[is.na(sy) | is.na(tot)] <- eprev[wave]
+ prev <- inprev.z[1,wave]*yfrac + inprev.z[2,wave]*(1-yfrac)
+ bigb3 <- 1-phi*beta*prev ;
+ bigc3 <- 1-phi*beta*prev*theta ;
+ nu <- (dataset[,"nrai"]-dataset[,"nrai.prot"])
+ np <- (dataset[,"nrai.prot"])
+ ninf <- bigb3^nu * bigc3^np * exp(-tau*beta*prev*(nu+theta*np))
+ ninfm <- 1-ninf
+ xx<-(dataset[,"hivstatus"])
+ ll<-sum(log(ninf*(1-xx)+ninfm*xx))
+ ll
+ }
|
Let's go through this line by line.
simlik.1<-function(inpars,dataset,inprev.z,eprev) {
wave <- as.integer(dataset[1,"studywave"])
beta <- inpars[wave-2] ;
theta <- inpars[5] ;
|
Here, we simply have the name of the function simlik.1 and
the formal parameter list. The first line takes the study wave
variable out of the first data point; the assumption is that this
variable is the same for every record. A more efficient way to do
this would be to simply not have this variable at all, and just
pass a single integer wave as the argument. Another
solution might be
simlik.1<-function(inpars,dataset,inprev.z,eprev,wave=as.integer(dataset[1,"studywave"]) {
beta <- inpars[wave-2] ;
theta <- inpars[5] ;
|
making wave an optional argument which would be determined
from the data set unless it is specified. After that, we simply
determine which beta we want; only one beta applies
to each data set. Study period 3 of the YMHS corresponds to the first
beta, so we have to subtract the 2 given our conventions; then
theta is always the fifth element. We could also have
chosen to use named elements as well, for greater robustness and
clarity (but at the cost of slower run time).
phi<-pmin(1,dataset[,"frac.yrs.since.test"])
tau<-pmax(1,as.vector(dataset[,"frac.yrs.since.test"]))-1
|
Here, we need something different. Remember that phi has to
be the chance a reported partnership happened since the test; if
the reporting period is greater than 12 months, then this is always 1.
What we want to do is take each observed frac.yrs.since.test
and make a new vector, with the observed fraction if it is less than
1, and simply 1 if it is greater. To do this we need a vectorized
kind of minimum function; we don't want the minimum of all the
elements in a vector, but an elementwise minimum. For instance:
> z <- c(0.2, 0.3, 1.5, 0.6)
> min(1,z)
[1] 0.2
> pmin(1,z)
[1] 0.2 0.3 1.0 0.6
|
We use the function pmin for this, and
as usual if the two vectors given as arguments are of different
lengths, the shorter one is promoted by duplication as many times as
necessary to match the length of the longer. And then similarly, to
determine tau, the amount of time beyond 12 months since the
last HIV test for this patient. Here, we use pmax, and
subtract 1; for example:
> z <- c(0.2, 0.3, 1.5, 0.6)
> max(1,z)
[1] 1.5
> pmax(1,z)
[1] 1.0 1.0 1.5 1.0
> pmax(1,z)-1
[1] 0.0 0.0 0.5 0.0
|
The next lines simply compute a vector of the fractions of
partners under 30 for each person, imputing missing values from the
overall fraction for that wave. Note that eprev must be
a vector; the i-th element of the eprev-vector is the
young partner fraction for study period (wave) i. Finally, the
weighted prevalence of infection for the partners of each person
is computed in the last line, and called prev.
sy <- as.vector(dataset[,"nyoung"])
tot <- as.vector(dataset[,"ntot"])
yfrac <- sy/tot
yfrac[is.na(sy) | is.na(tot)] <- eprev[wave]
prev <- inprev.z[1,wave]*yfrac + inprev.z[2,wave]*(1-yfrac)
|
Finally, we compute the log-likelihood as follows. We compute (as
vectors) the escape probabilities for each individual, for
protected partnerships (bigc) and unprotected partnerships
(bigb). We then get the number of unprotected partnerships
(nu) and protected partnerships (np); note that
these are all vectors. Finally, we compute (again, as a vector, a
value for each subject) the noninfection probability for that wave
as ninf, and the infection probability ninfm as
one minus that. The vector of values xx is the seroconversion
status for each subject. When the subject is infected, the infection
probability is computed; when the subject is not infected, the noninfection
probability is used. This is calculated in the
ll line; the log of each element is calculated,
and then all these values summed up. This reflects the
assumption that the subjects are independent, so the total likelihood
is the product of the likelihood of each individual subject; the
log of the product is then the sum of the logs. And this value
is what is returned from the function.
bigb <- 1-phi*beta*prev ;
bigc <- 1-phi*beta*prev*theta ;
nu <- (dataset[,"nrai"]-dataset[,"nrai.prot"])
np <- (dataset[,"nrai.prot"])
ninf <- bigb^nu * bigc^np * exp(-tau*beta*prev*(nu+theta*np))
ninfm <- 1-ninf
xx<-(dataset[,"hivstatus"])
ll<-sum(log(ninf*(1-xx)+ninfm*xx))
ll
}
|
Next, how do we use this function? This computes the log-likelihood for
one data set; there are four data sets. We compute the overall
log-likelihood in a simple way:
> loglik <- function(inpars,d3,d4,d5,d6,inprev.zz,eprev) {
+ if (any(inpars<0) | any(inpars>1)) {
+ ans <- -1e200
+ } else {
+ ans <- simlik.1(inpars,d3,inprev.zz,eprev) +
+ simlik.1(inpars,d4,inprev.zz,eprev) +
+ simlik.1(inpars,d5,inprev.zz,eprev) +
+ simlik.1(inpars,d6,inprev.zz,eprev)
+ }
+ ans
+ }
|
This takes the inpars (which has the same format as
we discussed, i.e. four period-specific values of beta, and
then theta), four data sets, a matrix of prevalences
inprev.zz, and the vector of young partnership fractions
eprev. All that happens is that if any of the probabilities
are out of the allowable range between zero and one, a very large
negative value is returned for the log-likelihood; otherwise the
likelihood is computed for each data set and all added up. Notice
this assumes independence between data sets of each observation. The
value -1e200 was found by experimentation; -Inf
would have been much better, but some optimizers complain if a non-numeric value is returned.
Then we can simply perform the optimization using
> answer <- optim(c(0.11,0.11,0.056,0.043,0.04),
+ function(arg)loglik(arg,data3to4final,data4to5final,data5to6final,data6to7final,inprev.1,eprev)
+ control=list(fnscale=-1))
|
The R builtin function optim performs multidimensional
optimization using a variety of methods. We call it for our
likelihood function, after substituting in all the data sets,
desired input prevalences, and so forth. The function optim
is quite complicated and has many options; because we're trying to
find a maximum, we need to add the incantation control=list(fnscale=-1) according to the manual. Such matters will be discussed in more
detail in the second semester; nonlinear optimization is never a
thing to be taken for granted.
> answer
$par
[1] 0.11750527 0.12412787 0.05520114 0.04388523 0.05430423
$value
[1] -97.31828
$counts
function gradient
462 NA
$convergence
[1] 0
$message
NULL
|
What is all this? We get back a list, and the element par
corresponds to the parameters that maximized the function, in this
case, our log-likelihood. We can see that the per-partnership
infectivities were about 0.118, 0.124, 0.055, and 0.043; in this
case I knew about what they should be and had the optimizer find
their exact location; you always want to have a good idea of where to
start looking for an optimum. The final parameter was the value
of the condom parameter, and this was about 0.054. We also get back
what the log-likelihood actually was at the maximum, about -97.31.
The remainder is information about the numerical performance of
the algorithm it chose (in this case, the default Nelder-Mead
downhill simplex).
What we next did was to constrain the first two betas to be the same,
and the last two, and solve for the resulting three values using
the optimizer. Finally we constrained all the betas to be ths same,
and found the best single value for it. The difference between these
two reflects how important it is to the log-likelihood to be able
to choose a different value early and late. If this constraint does
not matter much, then there is not much evidence that the infectivity
changed. We performed the likelihood ratio chi square test to
test the hypothesis of constant infectivity, given our assumptions.
One way of viewing these data is to construct a kind of profile likelihood
surface. We first back up and recall that the actual transmission
probability per partnership was the product of the prevalence and
the infectivity. These two are not identifiable, but their product can
be estimated. We can then compute the infectivity if we know the prevalence,
and this is in effect what is happening. Let's aggregate the first two
waves (study visits) and the last two, so that we have really three things
to estimate from the data here: transmission probability per contact for the
first two periods, for the last two, and also the condom protection factor.
Suppose that we just pick the transmission probabilities, and then with those
fixed, find the best value of the condom factor. Once we have the value of
the condom factor that maximizes the likelihood, we compute the value of the
likelihood given the assumed transmission probabilities and the estimated
condom value. Let's then plot this value of the (log) likelihood.
A contour plot looks like this:
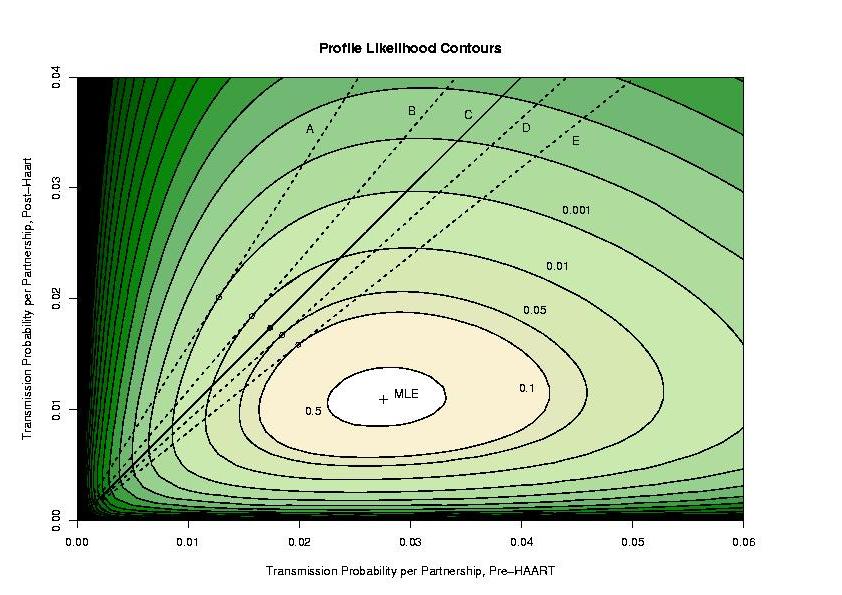
What does this mean? For each pair of transmission probabilities, we have
the best possible likelihood we can get (the largest value possible, found
by picking the condom protection factor to make it as big as possible). There
is a peak labeled with a cross, as you can see. That value represents the
maximum likelihood estimate of the transmission probabilities, and notice that
the value post-HAART is substantially lower than pre-HAART. We believe this
difference to be due to drugs, but this cannot be proven based on these data
alone (there is no control group and no possible way to assess the exposure
of the partners of the men in the study to the drugs.)
Now, suppose that we assume that the prevalence is the same pre- and post-HAART.
Let's now constrain the infectivity
to be the same. So with the prevalence the same and the infectivity the
same, we have to have the transmission probability be the same before and
after HAART. In other words, we have to stay on the 45-degree line on the
graph, shown on the figure in solid black. Given that, where is the
maximum likelihood estimate? It is at the black dot you see on it; if you
stay on that black like, that is the highest elevation you can get to and it
represents the best single (lumped) estimate of the transmission probability
per partnership. If the height you can get to is not far below the very
best (unconstrained) height (at the cross), then the constraint didn't make
much difference in a way. If that were to be the case, the data would
provide no evidence in favor of a difference. On the other hand, if having
to stay on the solid line dramatically lowered the best (greatest) likelihood
you could achieve, then that is evidence that you shouldn't have to stay
on the solid line: you would reject the hypothesis of constant infectivity
(given constant prevalence).
So which is it? We indicated contours that correspond to hypothesis rejection
at different alpha levels (by the likelihood ratio chi-square test), and that
is what you see on the graph. The solid dot (the maximum likelihood estimate
of the transmission probabilities given the hypothesis of no infectivity
difference, and constant prevalence) is between the 0.05 contour and the
0.01 contour, so the p-value is between 0.05 and 0.01. So at a level of 0.05,
we reject the hypothesis of constant infectivity.
Now, suppose that the prevalence increased post-HAART, since the death rate
due to AIDS decreased. Now, if we assume that the infectivity is the same
pre- and post-, what happens? We're assuming the prevalence after HAART
p2 is less than the prevalence
p1 pre-HAART. If we pick some infectivity
, we
can then compute the transmission probability per partnership before and
after HAART, respectively,
p1 and
p2. If we do this for a lot of different values
of the infectivity and plot the answers, we get a straight line, not at 45-degrees, but sloping upward at a steeper angle (since the prevalence has been
assumed to increase but the infectivity is the same, the actual
transmission probability per partnership just has to go up.) Given this
constraint, what is the best value? You find the highest point on the
transect, and see how much lower it is than the overall peak. In our
case, the steeper this slope, the lower it is, indicating that if the
prevalence actually increased, it is less plausible that the infectivity is
the same pre- and post-HAART. In particular, the dotted line A (at
to a rejection threshold of 0.001) corresponds to a 57.7% increase in
prevalence (but that is really too big to believe). The dotted line B
(at rejection threshold of 0.1) corresponds to a 17.2% increase in prevalence.
This goes the other way too. If we assume a prevalence decrease after
HAART (maybe people who were negative disproportionately and greatly
increased their risk behavior, diluting the positives...but there is no
evidence for this mechanism and we looked at our cohort at least). Now, the
solutions are constrained on a line with a shallower slope than 45-degrees.
The peak along these transects is higher, indicating that there would be
less evidence against the hypothesis of constant infectivity. In other words,
we would be partially explaining the findings by assuming a decrease
in prevalence. Assuming a 9.3% decline in prevalence takes us to the
rejection contour of 0.05; if we believe the prevalence decreased by more than
9.3% after HAART was introduced, we can no longer reject the hypothesis of
constant infectivity (line D). Finally the 0.10 rejection contour is
reached by assuming a 20.4% decline in prevalence.
I'll show how these were constructed in R in the future; this was a
somewhat numerically intensive calculation. Crude contour plots can be
made with the function contour which I invite you to explore.
Another thing that we did was to compute bootstrap resamples of
the data set each wave, and for each resampled data set, used the same
algorithm to find the best estimate. One problem, however, arises:
in a bootstrap resample, there might be no seroconverters at all,
and then the best estimate for the infectivity is zero, right on the
boundary. In these cases we had to eliminate this particular beta
from the optimization, since we already know what it is.
Another part of the final analysis was to assume a different per-partnership infectivity for a subset of the population, and compute simulated
infectivity data based on this assumption. We then fit the simulated
data and computed how often we falsely concluded there was a
infectivity drop due solely to frailty selection bias (using up the
people most likely to be infected, causing the average in the
cohort to fall).
In the future we'll add some of this code to this page; more details
and complete references can be found in the paper2.
In the next lecture, we'll look at a SARS preparedness analysis
programmed by Tomas Aragon.
1Osmond DH, Page K, Wiley J, Garrett K, Sheppard HW, Moss AR, Schrager L, Winkelstein W Jr. HIV infection in homosexual and bisexual men 18 to 29 years of age: the San Francisco Young Men's Health Study. American Journal of Public Health 84:1933-1937, 1994.
2 To appear.
3Grant RM, Wiley JA, Winkelstein W. Infectivity of the human immunodeficiency virus: estimates from a prospective
study of homosexual men. Journal of Infectious Diseases
156: 189-193, 1987.